Welcome to MetaGate!
MetaGate is a shiny-based R package that enables visualization and statistical analysis of large cytometry data sets through a simple web browser-based user interface. MetaGate was published in Patterns on May 13, 2024.
Please note that this R package, other R packages and R itself is free software that comes with absolutely no warranty. Please have a look at the License/citation section for more information.
Can MetaGate be useful for me?
If your data sets have large numbers of samples from well-estabilshed flow cytometry or CyTOF panels, MetaGate can help you organize and analyze your data in a simple and high-throughput way. Single- or multi-panel phenotypic and/or functional characterizations of patients samples are perfect MetaGate projects. MetaGate can also be useful for pooling data from separate experiments or quickly generating statistics and plots from simpler experiments.
If you are exploring novel cell subsets in a limited number of samples, other tools may be more useful. As a rule of thumb, if you are likely to report a p value from your flow cytometry or CyTOF experiment, MetaGate may be useful!
How does MetaGate work?
MetaGate is based on manual gating. This means that you still have to set and adjust gates for your samples, but it also makes your results easier to interpret and to integrate with existing data and knowledge. By letting you interactively define complex populations based on a limited number of gates, MetaGate can also help alleviate some of the frustration caused by very complex gating hierarchies.
After you have controlled your raw data and set the gates that you want in FlowJo or Cytobank, the gate file and FCS files are imported in MetaGate. Here, you can define populations based on your gates and select which channels you want to include. For all populations in all your samples, MetaGate then calculates population frequencies as well as mean, median and geometric mean values of all channels. By attaching meta data to the samples, groups can then very easily be compared using bar plots, heatmaps or volcano plots. You can also share your data with other users by sharing the relatively small .metagate files.
This document
This document was printed from the MetaGate website. Please go to https://metagate.malmberglab.com for the latest version.
Install MetaGate
The installation has two steps. First, you need to install R. Then, MetaGate can be installed through R.
Install or upgrade R
MetaGate is an R package, so before you start, make sure that you have R installed on your computer. If you have an old version, installing the most recent version might prevent some problems during installation. You can download R for free from the CRAN website:
- If you are using Mac, go to the R for Mac OS X page and download the file that ends with ".pkg" under "Latest release". When the download is completed, open the file and follow the installation instructions.
- If you are using Windows, go to the R for Windows page and click on the large "Download R x.x.x for Windows" link. When the download is completed, open the file and follow the installation instructions.
After completing the installation, you can open the R application to check if the installation was successful.
If you are using Mac, you might get error messages like "Setting LC_CTYPE failed, using "C"" or "You're using a non-UTF8 locale". If this is the case, run the following command in the R console and restart R: (Read more here)
system("defaults write org.R-project.R force.LANG en_US.UTF-8")
Install MetaGate
Now that you have R installed, MetaGate can be installed. Open an R console and enter the following commands to install MetaGate from the GitHub repository, along with required packages.
if (!require("remotes")) install.packages("remotes")
remotes::install_github("malmberglab/metagate")
During installation, R might ask you questions, like which mirror you want to use (selecting a mirror geographically close to you makes sense) or whether you want to create a personal library (which is usually a good idea if R asks). If you do not know how to respond or if any errors appear, help is very often to be found by googling. If installation seems to be done, you can now start MetaGate.
Launch MetaGate
To load the MetaGate package, enter the following code in R:
library(metagate)
If MetaGate was loaded successfully, it will output "Welcome to MetaGate!". You can now enter the following in R to start MetaGate:
run_metagate()
MetaGate will now launch in your web browser. If your web browser does not open automatically, open it manually and go to the address that was outputted by R.
You can now create your own project or get started using our sample project data.
To exit MetaGate, close the web browser, and hit the ESC key inside the R application. You can then quit R.
Sample data
If you want to try out the features of MetaGate without creating your own project, you can download the MetaGate file used to create all statistics and figures in the article Interactive Analysis of High-Dimensional Cytometry Data with Meta Data Integration (article in submission)
When you have downloaded the DLBCL.metagate file, launch MetaGate and select the DLBCL.metagate file in the Load project box.
FCS files can be downloaded from FlowRepository using accession code FR-FCM-Z6DF.
Create a project
Please follow the instructions provided inside the MetaGate application to create a new project and import your data in MetaGate. Once your project is created, you will have a stand-alone MetaGate file that can be used for visualization and statistical analysis. This file will be considerably smaller than the original FCS files, and while the project creation process can be computationally demanding and potentially time-consuming, the subsequent analysis and visualization will be much simpler and faster.
Some tips and tricks for data import
- If you are using Cytobank for gating, MetaGate will only use your gates, not your populations. Populations are defined inside MetaGate.
- If you are using FlowJo for gating, try to keep your gating hierarchy as simple as possible. See example here. You can then define populations based on these gates later inside MetaGate. It is not strictly necessary to do it this way (you can also import your very complex FlowJo gating hierarchy), but it may simplify the procedure.
Example: If you have a gating hierarchy with subsets for CD4+ and CD8+ T cells, gates set in one the subsets cannot be used in the other. However, gates set higher up in the gating hierarchy can be used in both subsets.
- After importing your FlowJo workspace file or Cytobank GatingML file, copy or move this to a safe place and avoid editing it. That way, you can go back and look at your gates at later stages in the analysis.
- In MetaGate, when you have defined your populations, export these as an Excel file by clicking on the "Export" button. If you want to import the same or similar data later, you can then import your population definitions.
- In MetaGate, make population names as simple as possible, as these will appear in plots. You can always access the definition for each population inside MetaGate.
- In the last step of the data import procedure, you can change the marker names. Make sure that these names are as simple as possible, as these will appear in your plots.
Note: If some marker names are spelled differently in your samples, these will be listed as multiple markers in the marker list. To solve this, change the names of these markers so that they become identical.
- Disabling the progress indicator in the last step of the import procedure can speed up the parsing. If you have a small number of FCS files (< 5-10), setting the number of cores to 1 can also potentially speed things up.
- The .metagates file contain all the information that MetaGate needs from the FCS files to make plots and statistical analyses. However, you cannot retrieve raw data from .metagate files or convert them to FCS files. Therefore, never delete your FCS files.
Analyze data in MetaGate
If no project is loaded, start by creating a project following the instructions above, or opening a saved project file by selecting this under Open saved project after launching MetaGate.
Remember to save your project if you want to keep your added meta data or groups. Click on the Save project button on the Project page. Please note that every time you save your project, a new project file will be downloaded, so no project files will be overwritten.
During analysis, yellow boxes with text might appear. These can provide valuable hints for getting further with your analysis. For some labels (e.g. Plot type or Mean bar), a pop-up with additional information will appear when you hold the cursor over it.
Add meta data to your project
In MetaGate, meta data is defined as information about samples. To be able to create groups and analyze your samples, you need to assign some meta data to each sample. This could for example be clinical data (e.g. diagnosis or treatment outcome) or experimental conditions (e.g. timepoint or stimulation type).
- Select the Meta data page in the main menu.
- Click on the Download Excel file template. An Excel (.xlsx) file will now be downloaded.
- Open the Excel file with Microsoft Excel, LibreOffice Calc or any other spreadsheet software.
- In this file, each row represents one sample, while each column represent a meta data variable (e.g. sex, diagnosis or timepoint). To create a new variable (e.g. sex), find an empty column, write "sex" in the top row, and fill in "male" or "female" for each of the samples. Samples can be identified by the FCS file name in the file column. Do not make the any changes to the file column.
Note: If you already have an Excel file (or similar) with a list of all your samples and related meta data, you can also use this as a starting point. To do this, create a new column called "file", and fill it with the FCS file names for each sample.
- Save the file (as .xlsx).
- Go back to MetaGate, click on the Upload Excel meta file and select the file you just saved.
- If your upload was successful, you will now see a list of all the meta data variables in the Summary tab of the Meta data page. The Samples tab shows you the meta data for each sample.
Note: By clicking on the Settings button on the Meta data page, you will be able to choose which meta data variables should be used for different purposes. By default, all variables are used for all purposes, and it is not essential to do any changes here. However, if you have a high number of samples or variables, making appropriate changes here could increase speed and usability of the software.
- You can now continue to creating groups. If you at any point during analysis want to change the meta data, just download a new template, make adjustments and upload.
Note: Remember to save your project after adding new meta data. You can do this by selecting the Project page in the main menu and clicking on the Save project button. Be aware that this will download a new project file, so your old file will not be updated.
Create groups based on your meta data
Now that you have assigned some meta data to each sample, you can create groups based on this meta data. Groups let you combine all your meta data variables to enable comparison of very specific selections of samples.
- Select the Groups page in the main menu.
- Find an empty row in the table, and start by giving your group a name by typing in the first column.
- Next, go to the second column to give your group a definition. Do this by selecting options for any of your variables.
Example: Say that your meta data variables are "sex" (with the options "female" and "male"), and "diagnosis" (with the options "AML", "ALL" and "CML"). Leaving the definition blank will include all samples in this group. Selecting only "sex: male" will include all samples from male patients in the group (regardless of diagnosis). If you select both "sex: male" and "diagnosis: AML", only samples from male AML patients will be included. Selecting "sex: male", "diagnosis: AML" and "diagnosis: ALL" will include samples from male patients with either AML or ALL.
- When some groups are defined, you can go ahead and create plots. You can go back and add, remove or change groups at any point during analysis.
Note: If you have spent some time creating groups, it might be worthwhile to save your project. You can do this by selecting the Project page in the main menu and clicking on the Save project button. Be aware that this will download a new project file, so your old file will not be updated.
Note: If you update the meta data, groups and plots will automatically be updated accordingly.
Create plots and statistics
There are five main ways of visualizing data in MetaGate: dot plots, heatmaps, volcano plots, sample clustering and readout correlations. Use the main menu to select between these. Each analysis method is briefly described below.
Dot plots
Dot plots let you analyze single readouts across multiple groups. The readout is selected in the left column. Select one or more groups at the top of the right column. Analysis options and statistical tests varies based on the number of groups you select.
Heatmap
Heatmaps are great for visualizing multiple readouts at the same time. Select readouts in the left column and click on the Add readouts button. Then, select the groups you want to include. If only one group is selected, the heatmap will show the values of each readout with readouts on one axis and populations on the other one. If two groups are selected, the same kind of plot will be showed, but values will instead indicate fold change (or any other comparison method you choose). If more groups are selected, all groups will be showed side by side.
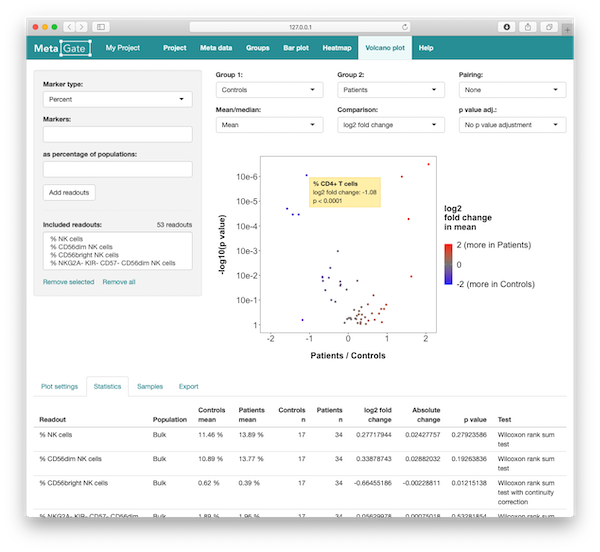
Volcano plots
Volcano plots let you compare multiple readouts between two groups by visualizing both the fold change and the statistical significance. Select readouts in the left column and click on the Add readouts button. Then, select the groups you want to compare in the drop-down menus at the top of the right column. In the volcano plot, each dot represent one readout. The X axis shows fold change (or absolute change), while the Y axis shows the negative base 10 logarithm of the p value.
About statistical testing
MetaGate performs statistical testing based on the number of groups you are comparing. Additionally, when testing mulitple readouts or groups, MetaGate lets you apply various p value adjustment methods. However, it is always the responsibility of the user to make sure that the choice and interpretation of statistical tests is appropriate for the data. You can see the p values and what test MetaGate used by selecting the Data table tab beneath the analysis. Click on the Download data as an Excel file button in the Export tab to look at this table in any spreadsheet software.
Glossary
Some of the technical terms used in MetaGate can be ambiguous. The definitions below are not intended as a suggestions for the correct interpretation of these terms in general, but rather as a guide to how these terms should be understood when using MetaGate.
- Project: A project can be stored in a .metagate file, and a .metagate file can only contain one project. Merging multiple projects will result in one project.
- Gate file: The FlowJo workspace file or the Cytobank GatingML file that was used to define gates when importing data.
- Sample: Equivalent to an FCS file. Analyzing one biological specimen using multiple panels will result in multiple samples.
- Readout: A metric that can be calculated for each sample. A readout is always defined by a marker and a population.
- Marker: A metric that can be calculated in a population. This could be a percentage or a mean, median or geometric mean value of a parameter. Examples: % CD4+ cells, % T cells, mean CD28 expression, median CD28 expression.
- Population: A set of events in sample, defined by one or more gates. Examples: T cells, CD4+, CD57+ CD8+ T cells.
- Bulk: A population created automatically by MetaGate. Includes all events in a sample.
- Parameter: A channel detected by the cytometer.
- Event: A sample contains multiple events, for which one or more parameters is recorded. An event typically represents a cell or a bead.
- Meta data: Categorical or numeric information assigned to each sample. The meta data consists of one categorical og numeric value per variable per sample.
- Variable: A column in the meta data file. Variables can be used to define groups, pair samples or to add annotations to plots. Examples: timepoint, diagnosis, disease stage.
- Group: A set of samples selected based on their meta data.
Questions
Install and launch
I have an old version of MetaGate. How can I upgrade?
To upgrade to the last version of MetaGate, simply run the installation procedure.
The installation fails
MetaGate depends on a large number of other R packages to run. When the installation fails, this is likely to be caused by problems installing some of these packages. Updating R to the most recent version could in some cases solve the problem. Otherwise, the error message outputted during installation may guide you to a solution.
Do I need an internet connection to run MetaGate?
MetaGate runs locally on your computer, and no internet connection is needed to open or use it. However, an internet connection is needed to install the software, as both R, MetaGate and required packages need to be downloaded. If you want to install MetaGate on a computer without an internet connection, the software can be downloaded on separate computer and transferred to the offline computer by other methods, but how to to this is not covered by this manual.
Do I need a powerful computer to run MetaGate?
Creating a new project and importing data requires a large number of calculations, and using a powerful computer can reduce the running time of this process. The downstream analysis (including meta data handling, plot generation and statistical analysis) does usually not require much computational power and should run smoothly on most computers.
Can I open multiple projects at the same time?
Yes, just open another tab or window in your web browser, and go to the same address (usually something like http://127.0.0.1:3838). Be aware that these tabs/windows will share the same "processing capacity", so if you are parsing/importing data in one of the windows, the other window may not respond.
Creating projects
Are all FCS files supported?
Only version 3 FCS files are supported. FCS files from some instruments may cause problems or not work at all. In this case, try importing the FCS files to FlowJo or Cytobank, set a gate, and export the events in this gate as new FCS files. We recommend to always gate away un-wanted events (like debris, dead cells, doublets), verify compensation, then export as new FCS files (with compensated values) and import these FCS files in FlowJo or Cytobank for gating. When importing data into MetaGate later, use the "new" FCS files.
Is compensation applied to my data?
If a compensation matrix is found inside the FCS file, this will be used to compensate values. However, new compensation matrices created in e.g. FlowJo will not be available for MetaGate, and some instruments may potentially add compensation matrices to the FCS files that MetaGate does not understand. Therefore, whenever working with fluorescent data, it is strongly recommended that you export new FCS files with only compensated values. That way, you can be certain that MetaGate uses the correct values.
Can I use multiple panels?
MetaGate will exclude any gates that are not set for all samples. If the difference between your panels is so that you want to set different gates, do the following:
- Gate the samples from each panel in separate FlowJo workspaces or Cytobank experiments.
- Create separate MetaGate projects for each panel and save the .metagate files.
- Merge the .metagate files by using the "Merge projects" feature in MetaGate. You will now have data from all panels in one MetaGate project.
If you do not need to set different gates in the different panels, you can import samples from all panels at once. In the "Select parameters" step of the import procedure, MetaGate will show you which parameters are found in which samples.
See also the question about panels in the "Analyze data" section below for more information about how to handle different panels during analysis.
I have misspelled some channel names. Can I correct that in MetaGate?
Yes! Let us say you have analyzed 40 samples with the same antibody panel, split on two experiments. In the first and second experiment, you labeled the FITC channel "PD–1–FITC" and "PD1–FITC", respectively. MetaGate will interpret these as two different parameters, and display them on separate rows in the "Choose parameters" step of the import procedure. To tell MetaGate that this is the same parameter, change the parameter names so that they are identical.
Can I define more populations than 256?
By default, 256 populations can be defined in MetaGate. If you want to increase this limit to 512, launch MetaGate using the following code:
run_metagate(populations = 512)
Be aware that setting a very high number may affect performance of the application.
What transformation should I select?
Transformation is only used for calculating mean, median and geometric mean values, and will not affect gating and population frequencies. Selecting "linear" will not transform the data at all. "Arcsinh" transformation is often used for mass cytometry data, while the "FlowJo logicle" can be used for flow cytometry data. Linear (no transformation) is usually a safe choice.
What is the transformation cofactor?
A transformation cofactor can be set when using arcsinh transformation and is a value that all values will be dived by before arcsinh transformation is applied: arcsinh(x / cofactor). A cofactor of 5 is quite common for mass cytometry data.
Analyzing data
The FCS file names are different from those shown in FlowJo
When an FCS file is created by e.g. a flow cytometer, its name is stored within the file (known as $FIL), in addition to being used as the name of the file itself in the file system (file name). FlowJo by default displays $FIL, while MetaGate always uses the file name. Therefore, if you manually change the names of the FCS files, or this is done by some software, samples will have different names in FlowJo and MetaGate.
Why does not MetaGate just show $FIL? To be able to identify samples, MetaGate requires all FCS files to have unique names. Sometimes, multiple samples may have the same $FIL, and changing it is difficult. This could for example happen when you debarcode CyTOF data. File names are usually unique (because the file system requires them to be if they are stored in the same folder), they are much easier to change.
Note: FlowJo can display the file name in addition to $FIL. In FlowJo, right-click on the header of the sample list and select "Edit columns...". From there, add "File Name" to the list of columns to display.
How can I define more groups than 20?
By default, 20 groups can be defined in MetaGate. To increase this number, use this code to launch MetaGate (replace "20" with the number of groups you want to use):
run_metagate(groups = 20)
Be aware that setting a very high number may affect performance of the application.
Some of my meta data variables are not available when I am defining groups
Make sure that these variables are selected in the "Group definition variables" field of the meta data settings. The meta data settings can be edited by clicking on the "Settings" button in the top-right corner of the Meta data page.
Note: The "file" variable is never available for group definitions. If you want to select single samples, create a new meta data variable for this purpose.
What is meant by "Bulk"?
"Bulk" is a special population created automatically by MetaGate that contains all events in a sample. In heatmaps and volcano plots, "Bulk" will be selected automatically if no population is selected. "Bulk" is sometimes omitted from readout names ("% T cells in Bulk" is displayed as "% T cells").
Why are some or all of the geometric mean values missing?
This is most likely caused by values below 0 in your data. MetaGate cannot calculate geometric means of negative values. Selecting a transformation method when importing the data can in some cases solve this.
How do I deal with multiple panels during analysis?
MetaGate lets you include data from different panels in one project. Please see the "Can I use multiple panels?" question above for instructions on how to create a MetaGate project with multiple panels.
When multiple panels are included in a project, problems can arise if the same readout is analyzed in more than one panel. For example, if you have gated on T cells in both panels of a two-panel project, T cell percentages will be included from both panels in your plots and statistical analyses. To avoid this, follow these instructions:
- Create a variable in your meta data that indicates which panel each sample belongs to.
Example: The variable could be named "panel" and have values like "T cell panel" and "Myeloid panel".
Note: If you created your project by merging multiple projects, MetaGate has already created a "gate_file" variable in your meta data. If the gate files corresponds perfectly to the different panels, there is no need to create a panel variable, as the "gate_file" variable can be used for this purpose.
- Go to the meta data settings. (Click on "Settings" in the top-right corner of the Meta data page.)
- Select your panel variable as "Panel variable" and click on "Apply changes".
For each readout, MetaGate will now display data from only one panel. You will be notified which panel was used for each readout during analysis. If you are analyzing multiple readouts at once (in a heatmap or volcano plot), different panels can be used for different readouts.
Note: When a readout is found in more than one panel, MetaGate chooses the panel for which the number of available data points is highest. If the number of data points is the same in multiple panels, MetaGate will choose the panel that appears first in the meta data table. If you want to specify which panel should be used, create a group where you specify the panel in the group definition.
Statistics
What do the asterisks (*) represent in the statistical analysis?
*, **, *** and **** indicate p values below or equal to 0.05, 0.01, 0.001 and 0.0001, respectively. To change this, use this code when launching MetaGate:
run_metagate(asterisk_limits = c(0.01, 0.001, 0.0001, 0.00001, 0.000001))
With this setting, *, **, ***, **** and ***** will indicate p values below or equal to 0.01, 0.001, 0.0001, 0.00001, 0.000001, respectively. To change back to default settings, restart MetaGate.
When comparing more than two groups, why is post-hoc comparisons not always performed?
In multiple-groups comparisons, post-hoc Dunn's test is only performed if the Kruskal-Wallis test gives a p value below or equal to 0.05. You can adjust this cut-off by using this code when launching MetaGate (replace "0.05" with the cut-off you want to use):
run_metagate(posthoc_limit = 0.05)
If the limit is set to 1, post-hoc analysis will always be performed.
What is a paired analysis, and how can I perform one?
In MetaGate, a paired analysis refers to a comparison of two groups that are related, and that you would normally test using a paired/one-sample statistical method. This could for example be samples from the same individual taken at different timepoints or under different conditions.
To be able to perform a paired analysis, MetaGate needs to know which samples should be linked to each other, and these samples need to share a common identifier supplied in the meta data.
Example: If your data consists of patient samples from two different timepoints, you need to add a variable in your meta data that contains a patient identifier, in addition to a variable specifying the sample timepoint. In the meta data settings (in the top-right corner of the Meta data page), set your patient identifier as a pairing variable. Then, create one group for each timepoint and compare these two groups in e.g. a bar plot. You can now select your patient identifier as a pairing variable. A paired statistical analysis will now be performed, and all incomplete pairs will be removed. If you select "Dot plot" as the plot type, the plot will show how which values are connected.
Some samples disappear when I run a paired analysis
If sample pairing is activated, MetaGate will remove all incomplete pairs.
Example: Let us say that you have analyzed paired patient samples from two timepoints. For some of the patients, samples were not available at one of the timepoints. These patients will then be removed from plots and statistical calculations when sample pairing is activated.
How do the p value adjustment methods work?
In MetaGate, p values are adjusted using the p.adjust function of the stats package in R. Please have a look at the documentation for p.adjust for details.
How should the box plots be interpreted?
In box plots, the middle horizontal line represents the median value, and hinges correspond to the 25th and 75th percentile. Whiskers range to the most extreme values, but no longer than 1.5 times the inter-quartile range. Data points outside that range are plotted individually as dots.
Citation and license
How to cite
Please cite the following article:
Ask EH, Tschan-Plessl A, Hoel HJ, Kolstad A, Holte H, Malmberg KJ. MetaGate: Interactive analysis of high-dimensional cytometry data with metadata integration. Patterns. 2024 May;100989. https://doi.org/10.1016/j.patter.2024.100989.
License
MetaGate is licensed under the GNU General Public License v3.0.
Source code
The full source code for MetaGate can be found in the MetaGate GitHub repository.